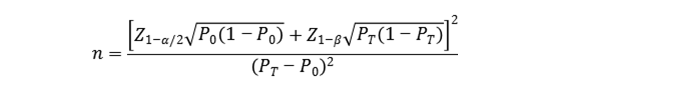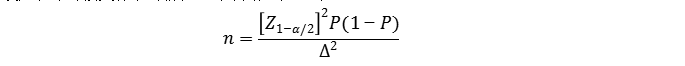四、临床试验质量管理 体外诊断试剂临床试验应符合《医疗器械临床试验质量管理规范》的相关要求,维护临床试验过程中受试者权益和安全,保证临床试验过程规范,结果真实、准确、完整和可追溯。临床试验质量管理应涵盖临床试验的全过程,包括临床试验的方案设计、实施、监查、稽查、检查,数据的采集、记录、保存、分析、总结和报告等。以下针对体外诊断试剂临床试验质量管理中需要特别关注的问题进行说明: (一)临床试验前管理 1.临床试验前,应确保产品设计已定型,完成试验体外诊断试剂的分析性能评估、阳性判断值或参考区间研究、质量检验以及风险受益分析等,且结果应能够支持该项临床试验。 2.临床试验中使用的试验体外诊断试剂按照医疗器械生产质量管理规范的相关要求生产且质量合格。 (二)临床试验开展 各临床试验机构原则上应当尽量同期开展临床试验,如在时间阶段上有较大的差异,应有合理的解释,确认采用了同一临床试验方案,并进行偏倚和中心效应分析。 (三)数据与记录 1. 在临床试验中,主要研究者应当确保将任何观察与发现均正确完整地予以记录。临床试验的源数据至少应当包括: 1.1所使用的试剂和仪器的信息,包括名称、规格/型号、批号/序列号、数量、接收日期、储存条件、使用情况及剩余试剂的处理等。 1.2受试者筛选入选记录、受试者基本信息(如性别、年龄、入组时间等)、临床诊疗信息、样本检验记录以及不良事件等。 1.3临床试验用样本来源、编号、保存、使用、留存、销毁等各环节的完整记录。 1.4 记录者的签名及日期。 2. 临床试验源数据不得随意更改;确需作更改时应当说明理由,签名并注明日期。 3. 样本管理及溯源:临床试验样本应由开展试验的临床试验机构提供,应具有唯一的可溯源编号,每一份样本应可溯源至唯一受试者(如有特殊情况应在方案和报告中说明)。 4. 临床试验数据应具有可追溯性,临床试验报告、病例报告表、临床试验数据表等文件中的数据均应一致且可以追溯至源文件。 (四)临床试验所需试剂管理 临床试验中试验体外诊断试剂、对比试剂及其配套使用的其他试剂(例如:核酸提取试剂等)和仪器、设备等的运输、使用、储存等,均应符合相关要求。 五、其他 1. 对于特定的体外诊断试剂,具体的临床试验方法、统计方法、样本量估算等可能有特定的要求,申办者应根据具体情况进行科学的选择和设计。如有相关产品的技术审查指导原则,应参考其中的相关要求。 2. 部分临床试验采用试验体外诊断试剂与核酸序列测定、GC-MS/MS等实验室检测参考方法进行比较研究,而这些方法并非临床常规检测技术,需要专门的设备仪器和试验条件,且大部分临床试验机构不具备相关检测条件。对于此类情况,申请人应尽可能选择具备相应条件的临床试验机构开展临床试验,确无检测条件的部分临床试验机构可将此部分测试委托给专门的测序机构、或具备一定检测资质的实验室进行检测,并对检测结果进行认可。产品注册申报时应提交临床试验机构与受委托机构的委托证明文件、对比方法的方法学研究资料和质控数据等。但不得委托申办者的实验室进行相关检测。由申办者直接委托的检测结果不得作为临床试验数据提交。
附件:样本量估算方法举例
附件 样本量估算方法举例
申办者应结合产品的具体特点、统计学分析模型等因素选择适当的样本量估算方法,并在方案中明确样本量确定的依据。同时应充分考虑可能的脱落剔除率等情况,合理设定样本量要求。 这里举例说明几种样本量估算方法。 一、定性检测的样本量估算举例 1.评价指标有确定的临床可接受标准时,需证明产品评价指标满足可接受标准要求。此时可采用单组目标值法样本量公式估算最低样本量。
公式中,n为样本量;Z1-α/2、Z1-β为显著性水平和把握度的标准正态分布的分数位,P0为评价指标的临床可接受标准,PT为试验体外诊断试剂评价指标预期值。 一般的,体外诊断试剂临床试验中,与已上市同类产品的对比试验均可根据临床需要设定适当的临床可接受标准,并采用上述公式进行最低样本量估算。 例:定性检测试剂临床试验,采用试验体外诊断试剂与已上市同类产品进行比较研究的方法,根据临床需求,阳性、阴性符合率应分别达到85%和90%,根据探索性试验结果,试验体外诊断试剂与对比试剂阳性、阴性符合率预期分别可达到90%和94%。 阳性组(n+)和阴性组(n-)最低样本量估计分别为:
根据以上估算,总样本例数预计为750例。 按照脱落剔除率10%,则应至少入组834例受试者,实际入组受试人群中,阳性组和阴性组样本例数应分别满足上述n+和n-的最低要求。 应注意,临床试验样本量除需满足上述统计学估算的最低样本量要求外,还应保证入组病例覆盖受试者的各种特征;如涉及不同样本类型,还需考虑不同样本类型的例数要求等。
2.对于临床试验的参数估计中只保证评价指标满足期望精度水平(置信区间的宽度一定),而不设定临床可接受标准的情况,可采用如下公式:
公式中n为样本量,Z1-α/2为置信度标准正态分布的分位数,P为评价指标预期值,Δ为P的允许误差大小。 应注意,P和Δ的取值应有充分依据,除非有特殊理由,否则不建议设置Δ>0.05,当预期值更高时还应考虑更优的精度。 采用上述公式,可根据灵敏度或特异度的预期值分别估算具有目标疾病状态的受试者(阳性)或不具有目标疾病状态的受试者(阴性)的样本量。
例如:某项标志物检测试剂用于相关疾病的辅助诊断,通过对已有资料进行分析得知,该检测试剂的灵敏度预期为85%,特异度预期为90%,临床试验采用试验体外诊断试剂与临床参考标准进行比较研究的方法,评价试验体外诊断试剂的临床性能。允许误差Δ取值0.05,则具有目标疾病状态的受试者(阳性)最低样本量(n+)估计为: 根据以上估算,总样本例数预计为334例。 按照脱落剔除率10%,则应至少入组371例受试者,实际入组受试人群中,具有目标疾病状态的受试者(阳性)和不具有目标疾病状态的受试者(阴性)样本例数应分别满足上述n+和n-的最低要求。 该临床试验中,除需满足最低样本量要求外,具有目标疾病状态的受试者(阳性)还应确保覆盖疾病的各种类型、不同疾病进程、不同疾病严重程度的病例;不具有目标疾病状态的受试者(阴性)则需涵盖临床特异度评价所需的各种受试者样本等。如果不同亚组人群预期灵敏度或特异度不同,还可能需要进行分层入组,并分别估算亚组样本量。 3. 需要注意的是,当评价指标P接近100%时,上述两种样本量估算方法可能不适用,应考虑选择更加适宜的方法进行样本量估算和统计学分析,如精确概率法等。 二、定量检测的样本量估算
对于定量检测,亦可针对适当的评价指标,选择适宜的统计学方法,进行样本量估算。临床试验方案中建议同时给出与所选定评价指标对应的临床可接受标准,并提供确定依据。 |