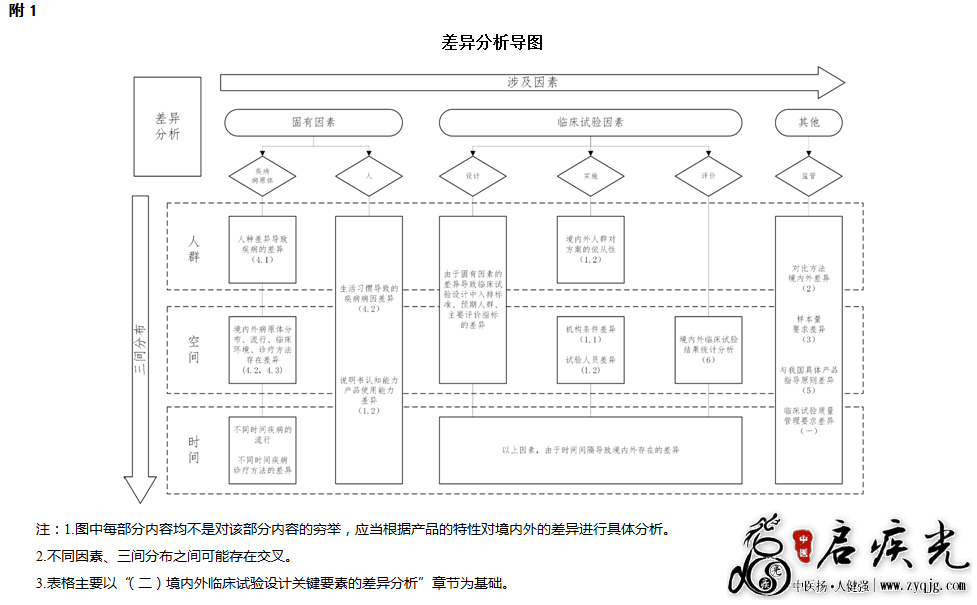
4.受试人群的差异 尽管境外临床试验数据支持申报产品在临床试验开展所在国家(地区)人群的使用,但部分产品由于境内外受试人群在人种、遗传信息、疾病特征或病原体流行情况等方面存在差异,造成境外临床试验受试人群不能代表境内受试人群的全部特征,从而导致境外临床试验数据无法充分支持申报产品在我国人群的使用。境内外受试人群的差异包括但不限于: 4.1不同人种遗传信息的差异 可能涉及该类差异的试剂主要包括人基因多态性试剂,如:药物作用靶点基因多态性试剂和药物代谢酶基因多态性试剂等。 例5:对于某些基因多态性试剂,同一多态性位点对于境内外人群可能具有不同的临床意义,导致境外基因多态性与药物使用相关性的临床证据无法直接外推至我国人群,如用于指导华法林用药剂量的VKORC1基因多态性试剂。不同患者华法林的用药剂量差异较大,这种差异受多种因素影响,其中VKORC1基因的单核苷酸多态性rs9923231(-1639 G>A)是主要因素之一。AA基因型患者的华法林平均使用剂量显著低于GA和GG基因型患者。该位点的多态性分布具有明显的种族差异,AA、GA和GG基因型在中国人群中的发生频率分别为79.7%、17.6%和2.7%,而在白种人群中的发生频率分别为14%、47%和39%。因此,该位点多态性对华法林剂量影响的权重在境内外存在显著差异。境外建立的基于该位点多态性的华法林剂量预测模型不能直接外推至我国人群。申请人应提供基于我国人群的临床证据,确定基于该位点多态性的华法林剂量预测模型。如有其他类似情况的药物作用靶点基因多态性试剂新产品申报,申请人应关注境内外基因多态性与药物使用的相关性是否具有差异。 例6:对于某些基因多态性试剂,同一多态性位点在境内外人群的发生频率可能存在差异,导致境外临床试验数据无法满足我国临床试验阳性例数的要求,如用于指导硝酸甘油用药的ALDH2基因多态性试剂。我国人群中ALDH2*2等位基因的携带率为30~50%,白种人和黑人几乎不携带,导致境外临床试验数据可能不含有ALDH2*2等位基因阳性病例。申请人应在我国境内或境外补充该等位基因的阳性病例,以满足我国临床试验有关阳性例数的要求。 4.2 疾病特征的差异 境内外疾病患病率、病原体感染率、疾病分型、疾病病因等的差异可能导致境内外临床试验数据的差异。 例7:疾病患病率和病原体感染率等的差异。疾病筛查类试剂受此因素影响较大,如人乳头瘤病毒(HPV)核酸筛查试剂。该类试剂用于鉴别具有高级别宫颈病变风险的人群,其临床试验数据受适用人群中宫颈癌患病率、HPV各亚型感染率及其导致宫颈癌病变的风险程度等多种因素的显著影响。境内外人群中宫颈癌的总体患病率和不同年龄段等亚组人群的患病率明显不同,并且不同病变程度宫颈癌等亚组人群中HPV亚型的感染率也明显不同。例如,在细胞学正常人群中,全球感染率排前五位的HPV亚型为HPV 16/52/31/53/18,我国则为HPV 52/16/58/33/18;在低级别宫颈上皮内瘤样变患者中,全球感染率排前五位的HPV亚型为HPV 16/52/51/31/53,我国则为HPV 16/18/58/52/33;在高级别宫颈上皮内瘤样变患者中,全球感染率排前五位的HPV亚型为HPV 16/52/31/58/33,我国则为HPV 16/18/58/52/33;在宫颈癌患者中,全球感染率排前五位的HPV亚型为HPV 16/18/45/33/58,我国则为HPV 16/18/31/52/58。因此,对于HPV核酸筛查试剂,上述差异可能造成境内外临床试验受试人群的明显差异,导致境内外临床试验数据的显著差异。针对该类筛查试剂,申请人原则上应在境内补充临床试验。否则,应提供充分证据,证明境外临床试验的受试人群可代表我国适用人群。 例8:疾病病因和疾病分型等的差异。对于食管癌和肝癌等多种肿瘤,不同人种在肿瘤病因、发病部位、病理特征和不同亚型分布等方面存在明显差异。例如,我国食管癌的主要病因是致癌性亚硝胺和某些真菌,而欧美食管癌的主要病因则为肥胖、胃食管反流和巴雷特食管;我国食管癌中鳞癌占95%,而欧美鳞癌只占30%;我国食管癌好发于上中段食管,而欧美食管癌多发于食管下 1/3 段。我国肝癌主要与HBV感染有关,而欧美则主要与HCV感染和酒精有关。另外,我国肝癌患者与欧美肝癌患者在流行病学、分子生物学特征、临床表现及分期上也具有明显差异。因此,对于预期用于食管癌或肝癌等的辅助诊断试剂,上述差异可能造成境内外临床试验受试人群的显著差异,导致境外临床试验数据不能外推至我国人群。申请人应结合产品具体特点(如:检测靶标等)进行充分分析,确认境外临床试验受试人群是否可代表我国适用人群。如有必要,应在境内补充临床试验。 4.3 受试人群中病原体流行情况的差异 针对病原体检测试剂,境内外受试人群中病原体流行情况的差异可能造成境外临床试验所验证的病原体不能代表境内病原体的全部特征,导致境外临床试验数据无法支持申报产品在我国人群的使用。 例9:境内外病原体流行基因型的差异。例如,乙型肝炎病毒(HBV)基因分型试剂,用于辅助医疗专业人员了解患者的乙型肝炎病毒基因型别,以便确定适当的治疗方法。HBV分为10种基因型(A~J),且HBV基因型的分布存在人种和地域差异。我国常见基因型为B和C型,部分地区存在少数D型,黑种人中常见基因型为A、E和D型,白种人常见基因型为A和D型,基因型F是美洲爱斯基摩人的优势基因型,基因型G主要分布在西方国家,基因型H在墨西哥的高加索和蒙古人种中占主导地位,而基因型I和J则主要在亚洲人中发现。因此,申请人应关注申报产品声称的可检出基因型及境外临床试验所验证的基因型是否涵盖我国流行基因型,各基因型数量是否满足我国技术审评的要求。如未涵盖或不满足,申请人应视情况在境外或境内补充临床试验。 例10:境内外病原体流行菌种的差异。例如,用于对血液样本中细菌和酵母菌等进行培养和定性检测的血培养瓶。境内外感染患者血液样本中的临床常见病原体种类可能不同,导致境外临床试验所检出的菌种不能完全覆盖我国临床常见的病原体菌种。另外,境内外临床常见菌种流行率可能存在差异,针对某些在境内流行率较高、但在境外流行率低的菌种,境外临床试验检出的菌种数量可能不足以验证申报产品对我国境内流行菌种的检测性能。申请人应关注申报产品声称的可检出的病原体菌种及境外临床试验所覆盖的菌种是否涵盖了我国常见病原体流行菌种,各菌种数量是否满足我国技术审评的要求。如未涵盖或不满足,申请人应视情况在境外或境内补充临床试验。 例11:境内外病原体耐药流行菌种的差异。例如,病原体耐药基因检测试剂。境外临床试验所检出的耐药菌种基因型可能不能完全覆盖我国临床常见的病原体耐药菌种基因型。例如,肺炎克雷伯菌对碳青霉烯类抗生素的耐药机制之一是产生碳青霉烯酶。碳青霉烯酶包括A、B和D三大类,每大类又分成各亚型。我国耐碳青霉烯类抗生素肺炎克雷伯菌主要产生KPC-2基因型碳青霉烯酶,西方国家则主要产生KPC-3基因型。申请人应关注境外临床试验所覆盖的耐药菌种基因型是否涵盖我国病原体常见耐药流行菌种基因型,各菌种基因型数量是否满足我国技术审评的要求。如未涵盖或不满足,申请人应视情况在境外或境内补充临床试验。 5.与我国具体产品指导原则的差异 申请人还应关注境外临床试验设计等是否满足我国相关产品指导原则的具体要求,如不满足,应详细阐明理由,并提供详细的研究资料,证明境外临床试验设计的合理性,必要时应在境内或境外补充临床试验。 例12:我国《结核分枝杆菌复合群耐药基因突变检测试剂注册技术审查指导原则》要求,结核分枝杆菌耐药基因突变检测试剂,如选择药敏表型作为临床参考标准,还需对所有耐药/阳性样本采用分子生物学方法进行验证,以明确引起耐药的具体突变类型,验证申报产品对耐药基因突变的检测性能。 6.其他可能的差异 对于某些产品,境内外临床试验的统计分析可能存在差异,导致境内外临床试验数据的差异。申请人应根据我国相关要求进行统计分析。 境内外临床试验数据的差异可能源于空间的差异、时间跨度的差异及人群的差异。申请人可参考本指导原则所述临床试验设计关键要素的思路,并结合附1“差异分析导图”所述路径,进行差异分析。 三、针对境内外差异的处理 申请人应根据产品具体情况,详细分析境内外临床试验可能存在的各种差异,确认境外临床试验数据是否满足我国注册申报临床试验的相关要求、是否能够充分支持申报产品在我国注册申报的预期用途。 如经分析发现,境外临床试验数据满足我国注册申报临床试验的相关要求,充分支持申报产品在我国注册申报的预期用途,则可将境外临床试验数据作为在我国注册申报的充分临床证据。 如经分析发现,境外临床试验数据无法完全满足我国注册申报临床试验的相关要求,不能充分支持申报产品在我国注册申报的预期用途,则可将境外临床试验数据作为在我国注册申报的部分临床证据,申请人应视情况在我国境内或境外补充临床试验,也可在我国境内按要求开展完整的临床试验。 四、使用境外临床试验数据的相关资料要求 (一)申请人应明确境外临床试验机构的名称及其所在国家(地区),境外临床试验数据的用途(如:用于申报产品的境外上市注册申报)。 (二)申请人应至少提交境外临床试验机构的伦理意见、临床试验方案和临床试验报告。伦理意见、临床试验方案和报告的提交形式、内容与签字签章等应满足境外临床试验所在国家(地区)临床试验质量管理的要求。申请人应提供完整的临床试验数据,不得筛选,临床试验报告应包含对完整临床试验数据的分析及结论。境外临床试验数据应真实、科学、可靠、并可追溯。 (三)申请人还应提交境内外临床试验的差异分析报告,应根据产品具体特点,综合分析各种可能涉及的差异及其对境内外临床试验数据的影响,并明确针对差异的处理情况。
|