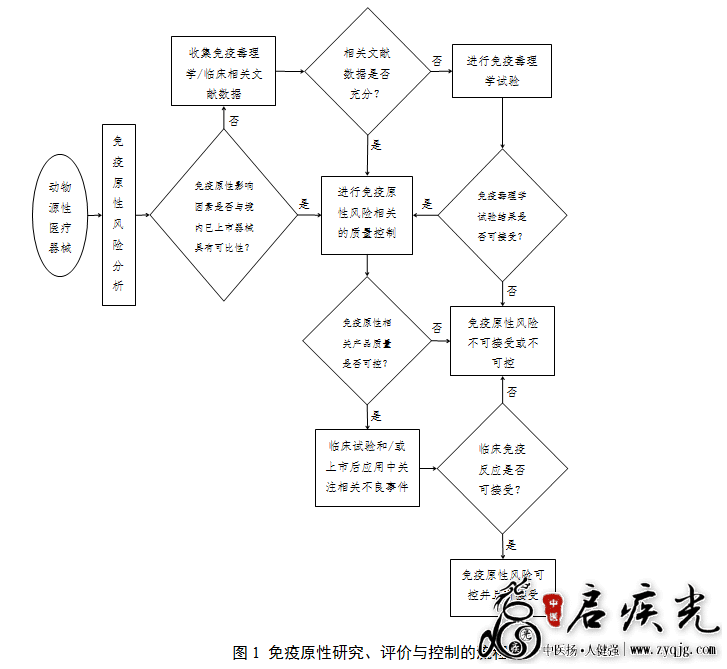
三、效果的判定 对于病毒去除/灭活效果的判断,应考虑同时达到以下两个要求: (一)去除/灭活降低系数的要求 病毒去除/灭活有效性验证的目的是为了确定生产工艺去除/灭活病毒的能力,因此需获得生产全过程中估计去除/灭活病毒的总降低系数。一般每种指示病毒的总降低系数为各步骤降低系数的总和。但是由于验证方法的局限性,如分步骤中指示病毒降低系数≤1 log,则不宜将其计算在总量中。在分析试验结果时需注意,如果将多步骤的去除/灭活病毒降低系数相加(特别是将去除/灭活效果不明显的步骤相加)或者将工艺过程中重复采用的同样或者类似去除/灭活机制形成的去除/灭活效果累加,可能会高估工艺实际能达到的效能。需考虑有效步骤对指示病毒的去除/灭活效果可能与实际生产工艺中使用的效果有一定偏差。 一般来说,医疗器械的生产过程中去除/灭活病毒的总降低系数宜达到6 logs以上(即病毒数量下降到进行去除/灭活前数量的百万分之一以下),并且原则上需至少有一个病毒去除/灭活步骤的降低系数达到4 logs以上(如因检测方法的灵敏度造成检测出的病毒降低系数接近但小于4 logs时,应盲传三代,如无病毒检出,亦可认为是有效地去除/灭活病毒步骤)。如果采用总降低系数达6 logs的病毒灭活/去除工艺将导致医疗器械产生不可接受的性能改变,则需要根据动物源性材料的来源、采集及处理过程控制情况以及对患者的风险/受益分析来判断其可接受性,但其单一去除/灭活病毒步骤的降低系数仍需达到4 logs以上。 即使验证研究证明了去除/灭活病毒工艺的有效性,这仅说明动物源性材料中残留病毒的感染性大幅度降低,但其数值永远不可能降至零。 (二)病毒灭活动力学要求 评价验证结果不能仅考虑病毒降低量,同时也要考虑病毒灭活动力学。需以作图的形式报告灭活动力学验证结果。如果指示病毒残留量很快降到最低检出限度值,则说明此方法灭活病毒效果较好;如果指示病毒灭活速率缓慢,在灭活结束时才达到最低检出限度值,则不能认为是一个有效的病毒灭活方法。 四、关于朊蛋白 由于目前尚难以采用致病性朊蛋白(如传染性海绵状脑病因子)的指示因子对去除朊蛋白的工艺进行验证,因此对牛、羊源性材料制品的传染性海绵状脑病安全性还主要是对源头进行控制。基于目前对朊蛋白去除/灭活工艺验证的认知程度,对于牛、羊源性医疗器械,可以接受按照本附录第一、二、三条阐述的原则所进行的病毒去除/灭活有效性验证。随着对朊蛋白研究水平的不断提高,相应的要求也将随时调整。 附录2 动物源性医疗器械免疫原性研究、评价与控制的原则 关于对动物源性医疗器械免疫原性的研究、评价与控制是建立在对产品免疫原性风险分析的基础上进行的,是动物源性医疗器械风险管理的一个组成部分。动物源性医疗器械免疫原性的研究、评价与控制资料主要包括:免疫原性毒理学/临床相关文献数据资料、免疫毒理学试验资料、免疫原性风险相关的质量控制资料以及免疫原性相关不良事件资料等。免疫原性研究、评价与控制的流程见图1。 一、免疫原性毒理学/临床相关文献数据 免疫原性毒理学/临床相关文献数据主要包括类似产品或材料作用于人体引发免疫应答的途径、发生免疫反应的种类、程度和可能性以及已报道的免疫毒理学数据等。引用文献时需注意文献数据的可靠性和与申报产品的相关性,并提供文献文本。 二、免疫毒理学试验 注册申请人可根据申报产品与已在境内上市产品在免疫原性影响因素(包括动物种类、取材组织、处理工艺原理、与人体接触方式等)上的可比性和免疫原性风险评价相关文献数据的充分性决定是否进行免疫毒理学试验。如申报产品免疫原性风险与已上市产品无可比性,且无充分的文献数据评价其免疫原性,则需进行免疫毒理学试验。免疫毒理学试验可按照YY/T 16886.20/ISO 10993-20进行。 注:在按照ISO 10993-20进行动物试验的免疫毒理学评价时宜充分考虑到动物种属对动物源性生物材料/医疗器械免疫反应的敏感性和特异性。 三、免疫原性风险相关的质量控制 即使申报产品与已上市产品免疫原性风险具有可比性或免疫原性评价的文献数据充分,甚至已通过免疫毒理学试验进行了免疫原性的评价,注册申请人也仍需进行免疫原性风险相关的质量控制。免疫原性风险相关的质量控制用于保证产品免疫原性降低工艺的稳定性,进而保证产品在批量生产后免疫原性风险持续可控。 建立免疫原性风险相关的质量控制,首先需建立能够反映免疫原性降低工艺稳定性的产品或中间品的性能指标(因体内试验不易操作且不易建立定量指标,故一般为通过体外试验建立的性能指标,如物理、化学指标),然后通过对这些性能指标进行验证和控制来实现对免疫原性降低工艺的稳定性以及批量生产产品免疫原性的控制。 注册申请人需结合动物取材组织中所含免疫原性物质的种类和数量、生产工艺中对免疫原性物质的处理方式、材料与人体接触方式等情况具体选择合适的控制方式。例如:对于通过提纯去除免疫原性物质的胶原产品,可通过杂蛋白的含量指标进行控制;对于通过脱细胞工艺去除免疫原性物质的产品,可通过残留细胞数量、残留DNA数量和/或残留α-Gal抗原的数量等指标进行控制;对于通过交联/固化方式使免疫原性物质失活或使抗原表位隐藏的产品,可通过表征交联/固化程度的指标进行控制。相关试验所涉及方法的国家/行业标准部分已发布(如YY/T 0606.25),部分正在研究和制定中。无论是否有相关标准,申请人均应按照已经过验证的方法进行试验。 四、免疫原性相关不良事件 经过了免疫原性的非临床评价及相关的质量控制之后,申请人还需在动物源性医疗器械的临床试验和/或上市后的临床应用中进一步关注与免疫反应相关的不良事件。
|