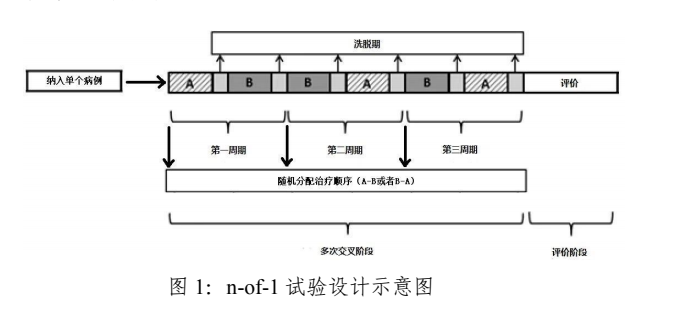
(二) 研究设计 通常情况下,随机对照试验通过随机分组最大限度地减少影响估计药物疗效的因素,因此研究结论的可靠性高,是评价药物疗效和安全性最有效、最准确的“金标准”。事实上,大多数已获批的罕见疾病药物均基于随机对照试验。对于常规的随机对照试验及可能适用于罕见疾病的剂量效应、延迟启动、随机撤药、交叉设计等设计方法,本指导原则不再详述,而主要阐述在常规随机对照试验中加入其他设计元素的方法(例如序贯设计、应答适应性设计、n-of-1 设计、适应性无缝设计、篮式设计、贝叶斯方法等)、单臂试验、真实世界研究等。若采用单臂试验设计、真实世界研究等作为注册申报的关键性证据,申办者应说明其合理性。 需要注意的是,任何一种研究设计都有其独特的优势和局限性。因此,实际药物开发中,申办者应根据研究目的和具体情形选择合适的设计并事先与监管机构沟通。 1. 序贯设计 序贯设计是指在控制总 I 类错误率的情况下,基于累积的数据进行期中分析,通过事先设定的合理的边界和样本量来判断疗效并决定试验是否继续。序贯设计适用于研究终点能够快速(相对于患者招募率)获得的临床试验。对于患者群体小、招募速度慢的罕见疾病临床试验,该方法可能适用。 2. 应答适应性设计 应答适应性设计是指新纳入受试者的随机分配概率根据已入组受试者的治疗结局而改变。这种设计的具体形式很多,常见的是“胜者优先”法,即根据已入组受试者的应答,如果一种治疗显示了更好的疗效,那么新纳入研究的患者更有可能在盲态下被分配到该治疗组中。此类设计可增加患者在潜在相对有效的治疗组中的暴露机会,同时可减少在剂量选择和确证性试验阶段的总体样本量。与序贯设计相同,该设计适用于能较快(相对于患者招募率)获得临床结局的试验。然而,这种设计不是基于随机分配概率固定的标准假设,且需注意盲态保持、统计分析等方面的问题。 3. n-of-1 设计 n-of-1 试验又被称为结构化的患者自身随机多交叉对照试验设计,简称自身多阶段随机对照试验。典型的 n-of-1 试验包括多个治疗周期(一般≥3),每个治疗周期内包括若干个阶段,受试者在每个阶段中接受某种治疗。第一个治疗周期内的治疗顺序(例如试验-对照、对照-试验)随机确定,之后每个治疗周期内的治疗顺序随机确定或使用系统性的平衡设计(例如,试验有两个组别,第一次随机确定的治疗顺序是对照-试验,后续治疗周期直接分配试验-对照、对照-试验的治疗顺序,并依此类推)确定。这种设计的主要目标是通过对同一受试者进行多周期的交叉治疗,观察该受试者对试验药物和对照药物的反应,从而为该受试者寻找最优治疗方案。当有多个受试者进行了相同设计的 n-of-1 试验时,可采用与交叉设计和 meta 分析相似的方式合并多个 n-of-1 试验的结果。一系列 n-of-1 试验通常能够更好地显示出疗效趋势。以 A、B 两种治疗 3 个周期为例,单个受试者的 n-of-1 试验设计示意图见图 1。
n-of-1 设计的优势在于利用患者自身对照设计,可以提高统计效率,减少样本量。同时能保证每个受试者都能得到阳性治疗。n-of-1 设计也有其局限性,例如,较适用于速效对症治疗和在治疗结束后迅速恢复到稳定基线值的疾病。对于疗程较长或起效较慢、以及自限性疾病,不宜使用 n-of-1设计。 需要注意的是,与一般的交叉设计类似,n-of-1 设计的前后不同阶段可能存在延滞效应。因此在试验的每个治疗阶段之间需考虑洗脱期。另外,受试者的随访时间比平行设计要长,因此受试者脱落的可能性较高。此外,研究设计还需要考虑治疗顺序的随机化和盲态的保持等问题。 4. 适应性无缝设计 适用于罕见疾病的适应性无缝设计主要是推断无缝设计,这种设计允许使用早期临床试验数据,在患者人群数量有限的情况下可能适用。例如,适应性 II/III 期推断无缝剂量选择的设计通常可以缩短由 II 期试验结束到 III 期试验开始时的时间间隔、减少试验总样本量、缩短试验时长等。另外,II 期入组的受试者有更长的随访时间,有利于更早观察到药物的长期疗效和安全性。在使用适应性无缝设计时需考虑控制总 I 类错误率、保持试验完整性(例如,防止期中分析结果被泄露导致影响研究者的后续操作以及受试者的入组)等问题。 5. 篮式设计 适用于罕见疾病的主方案设计主要是篮式设计。篮式设计旨在评估一种药物治疗具有同一种生物学特征的不同疾病类型的治疗效果,每一子方案针对一种或多种类型的疾病。 6. 贝叶斯方法 贝叶斯方法是将先验信息与试验的样本信息综合得出后验分布,再根据后验分布进行统计推断的方法。即利用先验信息校正研究结果。先验信息的来源包括但不限于历史研究、专家经验及无信息先验等。借用各种来源可靠的证据作为先验信息,可减少当前试验的样本量、缩短试验时长、提高检验效能,对于招募困难的罕见疾病可能适用。 为了获得充分的统计学证据,保证研究的质量、有效性和完整性,申办者应充分评估先验信息的合理性以及对统计学结论和最终结论可能带来的影响。建议使用其他合理的先验分布作为敏感性分析,以确保研究结论不会过分依赖先验信息。 7. 单臂试验 当罕见疾病患者数量极少、临床试验实施难度较大,尤其是当前缺乏有效治疗手段且危及生命的重大疾病,开展随机对照试验可能存在医学伦理风险,此时若考虑采用单臂试验设计,申办者需提供相应依据并阐明偏倚控制措施。 单臂试验通常采用外部对照,外部对照可以是目标值,也可以是外部的个体层面数据。 对于以目标值为对照的单臂试验,目标值的确定应有充分依据,它可以来源于前期研究(例如,meta 分析或某一个具有最佳参考意义的研究)的效应量,也可以是行业内广泛认可的效应量,以此作为试验组至少应取得的目标效应。以目标值为对照的单臂试验须在研究设计和实施过程中控制选择偏倚,保证入组患者的代表性和与历史对照的可比性,并在统计分析时考虑可能的偏倚(例如选择偏倚、幸存者偏倚等)。由于缺乏同期平行对照,其研究结果应谨慎解读。 对于以外部个体层面数据为对照的单臂试验,有平行对照和历史对照两种形式,鼓励采用平行对照。采用历史对照是真实世界研究的一种情形,如果采用历史对照,需事先对历史数据进行治理,当治理后的数据满足适用性要求后,才可开展相关研究。外部对照研究终点的选择应与试验组保持一致,如果某些临床终点的测量在外部对照与试验组并非完全一致,需事先评估其影响并在设计时提出应对措施。试验组的样本量估计仍需基于统计学假设或估计精度,外部对照的样本量需考虑匹配因素等方面,因此外部对照的样本量通常多于试验组。 8. 真实世界研究 真实世界研究是指针对预设的临床问题,在真实世界环境下收集与研究对象健康有关的数据(真实世界数据)或基于这些数据衍生的汇总数据,通过分析,获得药物的使用情况及潜在获益-风险的临床证据(真实世界证据)的研究过程。 若申办者考虑利用真实世界研究作为支持罕见疾病药物上市的关键证据,建议参照相关指导原则进行科学严谨的设计,并就方案、数据治理/管理计划、统计分析方法等与监管机构沟通并达成一致意见。
|