2、关注生物标志物的应用 生物标志物通常是指能被客观测量和评价,反映生理或病理过程,以及对暴露或治疗干预措施产生生物学效应的指标。罕见疾病患者人群小,难以开展大规模的临床试验,即使是开展了临床试验,也只能获得有限的有效性和安全性信息。由于多数罕见疾病的临床症状复杂多样,有些罕见疾病需要较长时间的药物干预才足以产生临床可识别的差异,因此,为了提高研发过程中识别药物干预产生的治疗效应或者安全性风险的敏感性,鼓励尽可能多的应用生物标志物,作为有限临床安全性和有效性数据的重要补充。例如,可利用安全性生物标志物,发现药物治疗中潜在的用药安全风险更高的患者;或利用药效学生物标志物,协助确定试验药物的合理给药方案,或开发可用于临床试验的替代终点等。 另一方面,还可以利用诊断性生物标志物,提高罕见疾病的确诊率,必要时需根据相关生物标志物开发伴随诊断。关于罕见疾病的伴随诊断开发可参考相关技术指导原则[1]。 3、积极应用定量药理学工具 基于罕见疾病受试者有限,患者年龄跨度大的特点,鼓励在研发过程中充分应用定量药理学工具,提高研发效率。例如,建立群体药代动力学-药效学模型,有助于科学、高效地确定试验药物在罕见疾病中的推荐剂量;有助于利用成人受试者数据外推不同年龄段儿童患者推荐剂量;有助于确定在特殊人群的推荐剂量等。 4、鼓励建立患者登记系统 罕见疾病单病种患者少,临床数据通常较为分散,收集和获取具有代表性的罕见疾病的临床数据存在难度,因此鼓励建立标准化患者登记系统。通过患者登记系统,有助于获得相对完整、准确的高质量临床数据,为统计和分析奠定良好基础,也为基于真实世界研究增加罕见疾病相关适应症提供可能。
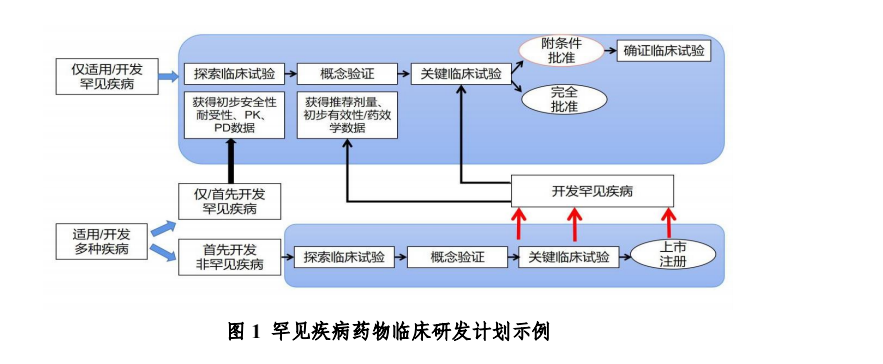
三、临床研发计划 根据罕见疾病药物的作用机制,可以分为两种情况:(1)只适用于目标罕见疾病;(2)同时适用于罕见疾病和非罕见疾病。 对于仅适用于罕见疾病的试验药物,通常需参考一般药物研发规律,开展早期探索研究,完成概念验证,确定推荐剂量、目标人群、获得初步有效性数据后,以此为基础开展关键研究,支持药物的上市。 在某些情况下,由于罕见疾病受试者有限,有时很难开展独立的概念验证研究,因此鼓励将关键研究分阶段开展,在第一阶段入组小样本量受试者,作为概念验证,并以此阶段结果为基础,对后续试验阶段进行调整,最终将第一阶段和后续研究阶段中,接受推荐剂量治疗的患者整体的有效性,作为支持上市的关键疗效数据。 对于适用于包括罕见疾病和非罕见疾病在内的多种疾病的药物,早期可以采用篮式试验设计,纳入多种疾病人群,并充分借鉴、利用在非罕见疾病中获得的临床数据,指导确定该药物在罕见疾病中的开发,根据在其他疾病所获得临床数据对罕见疾病适应症开发的指导价值,可考虑直接开展在罕见疾病适应症中的概念验证临床试验,或直接进入关键临床试验;当直接进入关键临床试验时,可参考前述适应性设计思路。 适用于包括罕见疾病和非罕见疾病在内的多种疾病的药物也可以选择首选开发罕见疾病,此时需参考情况“(1)只适用于目标罕见疾病”的情况进行开发。 罕见疾病复杂,病种繁多,药物研发难度较大,建议在开展罕见疾病药物研发工作时,对研发计划进行全盘考虑。鼓励申请人在研发过程中积极与监管机构就药物研发计划进行沟通交流。
四、临床试验设计 根据药物的作用机制,罕见疾病治疗药物一般可分为替代治疗及非替代治疗。替代治疗是指针对于人体内源性物质缺乏而导致的疾病,采用外源性物质予以补充的治疗方式;由于替代治疗补充的是人体所缺乏的内源性物质,因此通常替代治疗药物的作用机制明确,药物剂量也往往与内源物质的生理水平相关。非替代治疗是指替代疗法以外的其他干预性治疗,非替代治疗药物是通过干预疾病发生发展的途径中的一个或多个过程,或通过对非直接致病的旁路途径的干预,达到治疗疾病/缓解症状的治疗手段。非替代治疗相对于替代治疗而言,作用机制通常更为复杂,药物治疗剂量的探索也相对复杂。 罕见疾病的药物在不同研发阶段,需充分考虑罕见疾病发病率/患病率极低的特点,同时结合所研发药物的作用机制,合理设计临床试验。 (一)探索性研究阶段 1、研究人群的考虑 (1)健康受试者 通常健康受试者更为均一、干扰因素少,是早期探索研究(首次人体临床试验)的理想人群。尤其对于患者人群数量有限的罕见疾病,在健康受试者中获得药代动力学(pharmacokinetics,PK)、药效动力学(pharmacodynamics,PD)、初步 PK-PD 关系和初步安全性等信息十分重要。在拟定剂量范围内,药物作用机制预期不会对健康受试者造成严重危害,且非临床研究可支持在健康人群开展临床试验的前提下,可在健康成人受试者中开展首次人体临床试验。 (2)患者 对于不适合在健康受试者开展临床试验的药物,可在罕见疾病患者中开展,如果需要在儿童患者中开展研究的,原则上也应首先选择成人患者,在获得耐受性/安全性,药代动力学,药效动力学(如果可能)后,按照儿童药研发的一般原则[2],再逐步过渡到青少年和低龄儿童。 对于一些不是仅用于治疗罕见疾病的药物,即同时可作为其他疾病潜在治疗的药物,建议首先在非罕见疾病人群中开展首次人体临床试验。鼓励在早期研究中,采用篮式设计,根据药物作用机制,纳入包含目标罕见疾病人群在内的多种潜在适应症人群,收集在不同疾病中的安全性、药代动力学数据,为后期的研发提供依据。 对于极少数无法选择健康成年受试者,疾病人群仅为儿童,无成人患者可选择的特殊情况,也应在充分评估安全性风险的前提下审慎地考虑在儿童患者开展首次人体试验。 2、起始剂量的选择 药物起始剂量的确定应遵循药理毒理相关指导原则和技术要求[3]。 对于罕见疾病的替代治疗药物,由于对所缺乏的人体内源性物质的生理水平通常较为清晰,因此鼓励充分利用疾病的非临床研究和临床研究数据,建立替代治疗的药物剂量与所替代物质水平间的关系,在符合药理毒理相关技术指导原则对起始剂量要求,且安全可控的前提下,尽量选择接近于目标治疗剂量的水平作为起始剂量,以尽可能降低罕见疾病受试者的无效暴露,提高剂量探索研究的效率。 3、推荐剂量的确定 通常,推荐剂量是根据早期研究中药物的 PK、PD、安全性和初步有效性数据综合判断得出。在罕见疾病药物研发中,建议注重科学工具的使用。例如,采用模型引导的药物研发(model-informed drug development,MIDD)[4],开展群体药代动力学研究,建立药代动力学-药效学模型等,实现从健康人到患者,或从成人患者到儿童患者,或从其他疾病患者到目标罕见疾病患者的剂量外推。 对于作用机制明确的替代治疗,也可通过对 PK-PD 关系的充分研究,明确药物剂量-暴露量-效应关系,从而确定推荐剂量。 4、特殊人群用药 通常情况下,药物研发是先对成人适应症进行开发,之后再开展在儿童患者中的开发;就罕见疾病而言,按不同年龄段分别进行开发难度较大,且一些罕见疾病主要在儿童期开始发病之后进展至成年,儿童患者是重要的治疗人群,也是临床需求最强烈的人群;另一方面,通常药物会在研发到一定阶段,有的甚至在药物上市后,再开展肝/肾功能不全等特殊人群的研究,由于缺乏特殊人群的研究数据,在关键研究中常常将上述特殊人群排除。然而对于罕见疾病,由于疾病本身常会对患者肝、肾功能产生影响,因此,如果在临床试验中将上述特殊人群排除,入组患者就会更加困难,研究结果也不具有代表性,无法指导科学合理的临床用药来满足患者需求。 因此,在罕见疾病药物研发的早期阶段,建议适时开展特殊人群用药的研究,便于在后续关键临床试验中,可以尽可能的纳入更多、更广泛、更有代表性的罕见疾病患者。当明确不同人群(如老人、儿童、伴有肝/肾功能损害)的用法用量后,即使不同人群的用法用量不同,在关键临床试验中也可考虑一并纳入,从而提高研发效率。
|