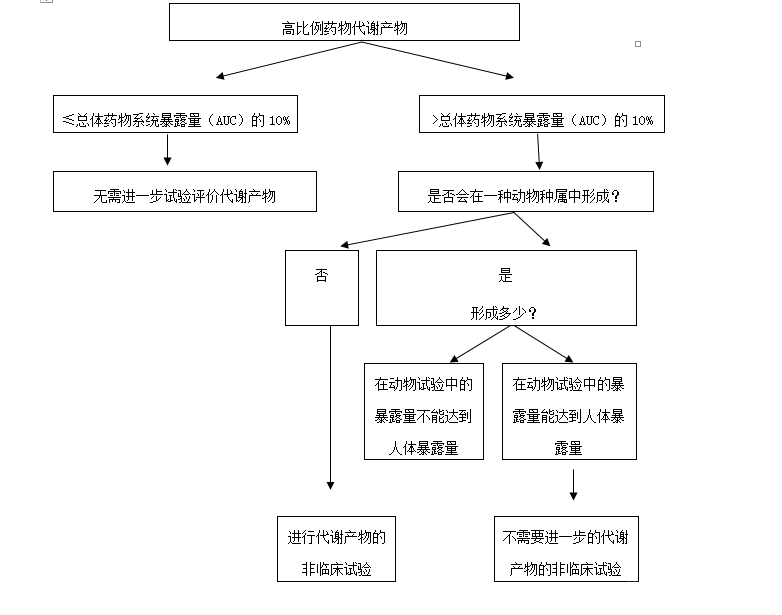
五、推荐的代谢产物的安全性试验 需要进行安全性评价的药物代谢产物的非临床试验应符合GLP。可能需要进行以下试验来评价高比例药物代谢产物的安全性。 (一)一般毒理试验 在一般毒理试验中,应评价高比例药物代谢产物的潜在毒性,进行代谢产物与其母体药物间的毒性比较。代谢产物直接给药的一般毒理试验的期限,可参考国内相关指导原则和ICH M3(支持药物进行人体试验需要的非临床安全性试验)技术指导原则。应在数倍于人体暴露量或者在至少与人体中检测到的暴露量相当水平下评价代谢产物的毒性。建议采用母体药物拟定的临床给药途径给药。但如果可以使高比例药物代谢产物达到足够的暴露量,也可以采用其他给药途径。如果临床给药途径为口服,确认代谢产物在胃液环境中的稳定性十分重要。为了保证高比例药物代谢产物的充分暴露,上述试验中需包括毒代动力学研究。 (二) 遗传毒性试验 需以一项检测点突变的体外试验和另外一项检测染色体畸变的试验来评价代谢产物的潜在遗传毒性。这些试验可参考SFDA《药物遗传毒性研究技术指导原则》。如果其中一项试验或两项试验结果是可疑和/或阳性时,可能需要进行完整的遗传毒性标准组合试验。 (三) 胚胎-胎仔发育毒性试验 如果药物拟定的用药人群包括有生育可能的妇女时,需进行代谢产物的胚胎-胎仔发育毒性试验。根据一般毒理试验和胚胎-胎仔发育毒性试验的结果,在具体问题具体分析的基础上,可能会要求进行其他生殖毒性试验。这些试验可参考SFDA《生殖毒性研究技术指导原则》。某些情况下,可仅在一种能形成该代谢产物的动物种属中进行胚胎-胎仔发育毒性试验。 (四)致癌性试验 药物连续用药至少6个月,或需间歇用药用于治疗慢性或周期性复发疾病,如果在母体药物的致癌性试验中不能对代谢产物的致癌性潜力进行充分评价,则应该进行代谢产物的致癌性试验。这些试验可参考SFDA《药物致癌试验必要性的技术指导原则》和国外的致癌性试验技术指导原则。 六、安全性研究的时间安排 需尽早确证人体中可能存在的高比例药物代谢产物,这可以为非临床试验的合理性提供证明,有助于阐明和制订临床试验计划,并避免药物开发进程的延迟。通常首先采用体外试验来预测动物和人体的药物代谢差异,然后根据人体和动物的代谢差异来考虑是否需要开展药物代谢产物的安全性试验。如果在体外代谢研究提示有人体的高比例药物代谢产物,可考虑开展动物和人体的同位素标记法的代谢研究,为开展高比例药物代谢产物的临床前安全性研究提供依据。 如果需要进行代谢产物的毒理学试验,应在大规模临床试验开始前完成这些试验,并向药品审评部门提交试验报告。 为了优化和加快研发用于包括肿瘤疾病在内的其它严重或危及生命疾病(如ALS、中风、HIV)的药物,对于那些具有重大治疗利益的药物,以及用于尚缺乏有效治疗手段疾病的药物,代谢产物的非临床试验的数量和类型可根据具体情况具体分析的原则进行调整。晚期癌症患者治疗时,这些代谢产物并不要求必需开展单独的毒理评价。此种情况下,申办人应联系相关药品审评部门进行讨论。 词汇表 代谢产物(Metabolite):母体药物经过I和/或II相代谢途径产生的物质。 药理活性代谢产物(Pharmacologically active metabolite):在靶受体具有药理学活性的代谢产物,其活性可能高于、等于或低于母体药物。 附录A 决策流程图
|