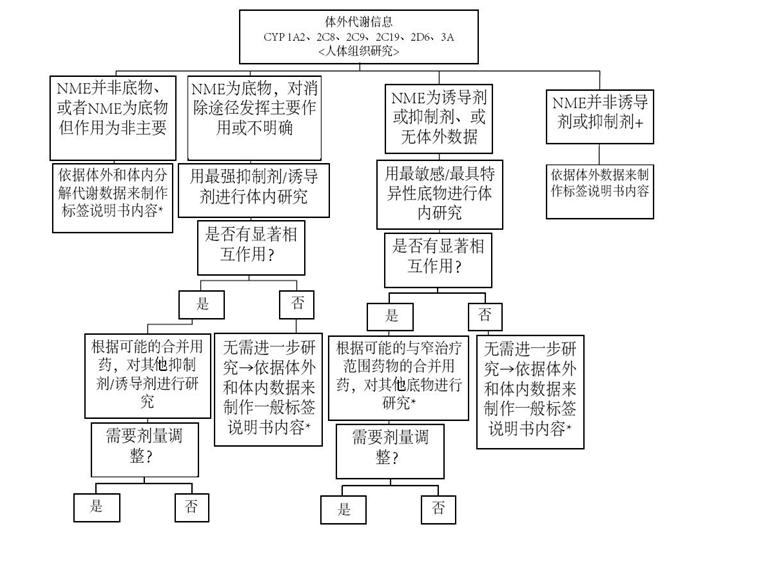
同样,如果体外研究结果表明所研究的药物对CYP1A2、CYP2C8、CYP2C9、CYP2C19、CYP2D6、或CYP3A代谢无抑制作用,那么就不需要进行相应体内的研究,即无需在这些酶抑制水平下进行研究药物与经这些酶消除的合用药物的体内相互作用研究。 CYP2D6酶未显示有可诱导性。最新数据显示CYP2C、CYP2B和ABCB1(P-gp)转运体与CYP3A具有协同诱导作用,CYP3A似乎对所有已知协同诱导物均敏感。因此,为了评价研究药物是否对CYP1A2、CYP2C8、CYP2C9、CYP2C19、或CYP3A有诱导作用,最初体外诱导评价可能仅包括CYP1A2和CYP3A。如果体外研究结果表明研究药物对CYP3A代谢不具有诱导作用,那么就不需要在这些酶诱导水平下进行研究药物与经CYP2C/CYP2B及CYP3A消除的合用药物的体内相互作用研究。 CYP2B6介导的药物相互作用是重要的相互作用。适当时,需进行基于该酶的相互作用的体外研究。其他CYP酶(包括CYP2A6和CYP2E1)被认为较少参与具有临床重要性的药物相互作用,只有在必要时,才考虑进行相关研究。
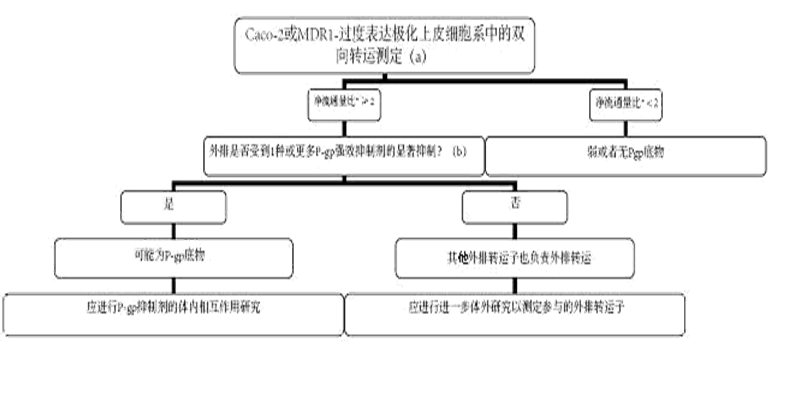
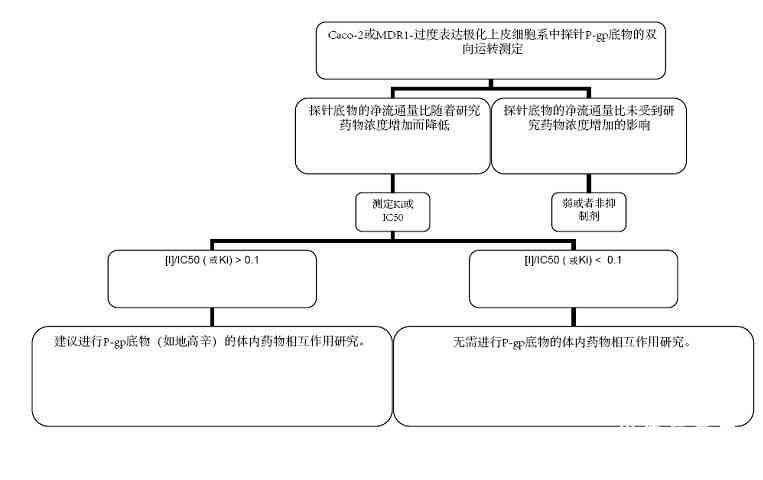
NME:新分子实体 * 补充的人群药代动力学分析结果可以帮助进行整体评价。 + 测定NME是否为特殊CYP酶的抑制剂 或诱导剂 的标准;鸡尾酒研究阴性结果将排除测定NME是否为特殊CYP酶抑制剂或诱导剂的进一步研究 。 图 2、3提供了根据体外评价结果而对体内P-gp相互作用研究的决策树作为参考。
* Caco-2细胞的净流通量比计算公式为(通透性app, B-A/通透性app, A-B);对于MDR1-过度表达细胞系,净流通量比计算公式为(通透性app, B-A/通透性app, A-b)mdr1与(通透性app, B-A/通透性app, A-B)野生型的比值。 (a) 合格系统产生的探针底物的下一个净流通量比应与文献报告值相似。研究药物的净流通量比>2是需要进一步评价的阳性信号。注意:考虑到此值太宽泛,因此会产生过多阳性结果的担忧。还可以使用%值(研究药物相对探针底物(如地高辛)的净流通量)。 (b) 流通量比值显著减少(> 50%)或接近一致。
* Caco-2细胞的净流通量比的计算公式为(通透性app,
B-A/通透性app, A-B);对于MDR1-过度表达细胞系,净流通量比的计算公式为(通透性app, B-A/通透性app, A-B)MDR1与(通透性app, B-A/通透性app, A-B)野生型的比值。[I]代表给予最高推荐临床剂量后、总药物(结合加未结合)的平均稳态Cmax值。 通常在药物研发早期阶段进行适当设计的药代动力学临床研究,可提供有关代谢消除的途径、对总体消除的贡献和代谢相关的药物相互作用的重要信息。结合从体外研究获得的信息,这些体内临床研究可成为药物产品说明书陈述的主要基础,并有助于免除进一步的药物相互作用研究。
对大规模临床研究中,通过稀疏或密集采集血样所获得的数据进行群体药动学分析,这种分析对于探讨已知或新发现的相互作用的临床重要性,以及对于提出剂量调整的建议都可能非常有价值。如果通过临床研究数据分析检测到药物相互作用引起药物暴露量的重要变化,这些分析可以为相互作用提供参考信息,有时可以得到结论。群体药代动力学评价有可能发现非预期的药物相互作用。当存在早期的证据以及作用机制的数据时,群体药代分析还可以提供不存在某种药物相互作用的进一步证据。但是,如果专门设计用以评估药物相互作用的体外或体内研究的信息强烈提示存在某种相互作用,那么群体药代分析不太可能用以证明不存在这种相互作用。群体药动学研究的试验流程和样品采集均应经过严密设计,才能从中获得最多的信息。
|