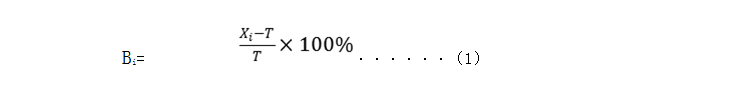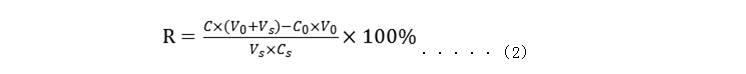(四)分析性能评估资料 申请人应当提交对申报试剂进行性能验证的研究资料,对于每项分析性能的评价都应包括具体的研究项目、试验设计、研究方法、可接受标准、试验数据、统计方法(如有)、研究结论等详细资料。性能评估时建议将试剂和所选用的校准品、质控品作为一个整体进行评价,评估整个系统的性能是否符合要求。有关分析性能验证的背景信息也应在申报资料中有所体现,包括试验地点、适用仪器(名称、型号、校准有效期)、试剂材料(名称、规格、批号、储存温度、有效期)及所选用的校准品和质控品、临床样本来源等。 对于本试剂(盒),建议着重对以下分析性能进行研究: 1.准确度 对测量准确度的评价依次包括:与国家标准品(和/或国际标准品)的相对偏差、回收试验等方法,申请人可根据实际情况选择以下方法的一项或几项进行研究。优先采用与国家(国际)标准品的比对研究。 1.1相对偏差法 如有相应国家(国际)标准品,则使用国家(国际)标准品进行验证,重点观察对相应标准品检测结果的偏差情况。申报试剂实测值与相应国家(国际)标准品的相对偏差应不超过±15%。 采用可用于评价常规方法的参考物质/有证参考物质(CRM)对试剂(盒)进行测试,重复检测3次,取测试结果记为(Xi),按公式(1)计算相对偏差(Bi)。实测值与标示值的相对偏差应在±15%范围内,如果3次结果都符合要求,即判为合格。如果大于等于2次的结果不合格,即判为不合格。如果有1次结果不符合要求,则应重新连续测试20次,并分别按照公式(1)计算相对偏差,如果大于等于19次测试的结果符合要求,则准确度判定合格。
式中: Xi—测试浓度; T—标定浓度; Bi—相对偏差。 注:首选国家参考物质,如无国家参考物质可选用国际参考物质。 1.2回收试验 用于评估定量检测方法准确测定待测分析物的能力,结果用回收率表示,是评估准确度的方法之一。 将已知浓度的高水平待测物(A)加入到低浓度的血清(或其他体液成分)B中,所加待测物A 与血清(或其他体液成分)B之间的体积比例为不大于1∶9,各重复检测3次,取平均值。可参考公式(2)计算回收率,25-羟基维生素D试剂盒各样本回收率及平均回收率应在[85%, 115%]范围间。
式中: R—回收率; C—向B液中加入A 液后的检测浓度的平均值; V0—B液体积; Vs—A 液体积; C0—B液浓度的平均值; Cs—A 液浓度。 回收试验注意事项: 1.2.1所加待测物A 与血清(或其他体液成分)B之间的体积比例应不会产生基质的变化,一般在样本体积的10%以内并且保证在加样过程中的取样准确度; 1.2.2保证总浓度在方法分析测量范围内,尽量使加入待测物后样本中的被测物浓度达到参考值; 1.2.3可通过稀释标准物制备不同高浓度的待测物以得到不同浓度的回收样本,标准物的浓度应该足够高; 1.2.4为减少基质效应,尽量采用与临床待测样本接近的基质,如血清(或其他体液成分)。 1.3方法学比对 采用参考方法或国内/国际普遍认为质量较好的已上市同类试剂作为比对方法,与拟申报试剂同时检测一批临床样品(至少40例样本),40例样本为在检测浓度范围内不同浓度的人源样品,且尽可能均匀分布。 从测定结果间的差异了解拟申报试剂与参比方法间的偏倚。如偏倚(参考值处)在企业规定的允许误差范围内,说明两检测系统对病人标本测定结果基本相符,对同一份临床样本的医学解释,拟申报试剂与对比方法相比不会产生显著差异结果。 在实施方法学比对前,应分别对拟申报试剂和比对试剂进行初步评估,只有在确认两者都分别符合各自相关的质量标准后方可进行方法学比对。方法学比对时应注意质量控制、样本类型、浓度分布范围并对结果进行合理的统计学分析。 建议与具有相同溯源性的分析系统作比对,相关系数(r)及对应斜率、相对偏差/绝对偏差应符合申请人的内部要求,并提供制定该要求的相关依据。 方法学比对注意事项: 1.3.1样本贮存时间及条件由被测组分的稳定性而定,尽可能避免使用贮存的样品; 1.3.2样品应来自于患者,并且此患者的疾病对于被测组分的影响应明确,尽量不使用含有干扰此方法的组分或条件的样本; 1.3.3分析浓度尽可能在报告的浓度范围内均匀分布。 2.空白限、检出限 申请人可参考国际或国内有关体外诊断产品性能评估的文件确定25-羟基维生素D试剂盒的空白限、检出限及参考区间等相关信息。25-羟基维生素D试剂盒(标记免疫分析法)产品检出限应不高于8.1ng/mL。 空白限:一般由多个空白样本的检测结果,经计算获得。当前阶段,企业申报的25-羟基维生素D试剂盒(标记免疫分析法)均采用竞争法。针对竞争法原理,可使用检测零浓度校准品或样本稀释液的方法进行。例如重复测定20次,根据测量结果的平均值(M)和标准差(SD),计算M-2SD及其对应的浓度值,结果应符合规定要求。 检出限:一般由多个低浓度(含有分析物)样本的检测结果,结合空白限进行计算获得,应根据具体产品的原理、检测结果差异和分布,选择合适的模型和分析方法(非参数/参数数据)进行计算。 空白限及检出限的验证:对5份浓度近似检出限的低值样本进行检测,每份样本检测5次,对检测结果按大小进行排序。低于申请人所确定的空白限数值的检测结果的数量应小于或等于3个;且无高于申请人提供的参考区间下限的检测结果的数值。此时,可认为申请人提供的空白限和检出限的设置基本合理。 检出限注意事项:空白样本应不含被测物,但其基质应与待测定常规样本相同。如空白样本难以得到,可采用去除维生素D的5%牛血清或人血清白蛋白溶液,或根据测定项目选用相应基质的样本,但应注意将基质效应减至最小。
|