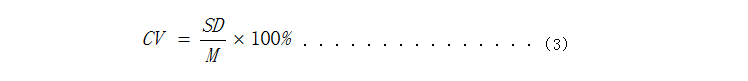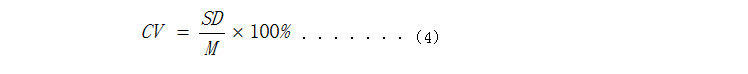3.线性 建立试剂线性范围所用的样本基质应与临床样本相似,但不可采用含有对测定方法具有明确干扰作用物质的样本。理想的样本为分析物浓度接近预期测定上限的混合人血清(或其他人源样本),且应充分考虑多倍稀释对样本基质的影响。建立一种定量测定方法的线性范围时,需在预期测定范围内选择7~11个浓度水平(建议采用梯度稀释)。例如,将预期测定范围加宽至130%,在此范围内选择更多的浓度水平,然后依据实验结果逐渐减少数据点直至表现出线性关系,可发现最宽的线性范围。 验证线性范围时可选择5~7个浓度水平。所选用的浓度水平应可覆盖整个预期测定范围并包括与临床有关的重要评价浓度,如最小测定浓度或线性范围的最低限、不同的参考值、最大测定浓度或线性范围的高限等。采用申报试剂对每个浓度至少重复测试3次,计算平均值,将结果平均值和稀释比例用最小二乘法进行线性拟合,计算线性相关系数r,线性范围应覆盖[8.1, 70.0] ng/mL,在申请人宣称的线性范围内,相关系数r应≥0.99。 4.重复性 测量重复性的评估应至少包括2个浓度水平的样本,两个浓度都应在申报试剂的测量范围内,建议采用人源样本或与人源样本基质接近的样本进行试验。当2个浓度的重复性有显著差异时,建议增加为三个浓度。 采用同一批试剂盒对选择浓度在[8, 20] ng/mL和[30, 50] ng/mL的样本各重复测试10次,计算10次测定浓度结果的平均值M和标准差SD,根据式(3)计算变异系数(CV)应不大于10%。
式中: CV—变异系数; SD—10次测量结果的标准差; M—10次测量结果的平均值。 5.批间差 用3个批号的试剂(盒)分别测试同一浓度为[30, 50] ng/mL的样本,各重复测定10次,计算30次测定浓度结果的平均值M和标准差SD,根据式(4)得到变异系数(CV)应≤15%。
式中: CV—变异系数; SD—30次测量结果的标准差; M—30次测量结果的平均值。 6.分析特异性 推荐参考《WS/T416-2013 干扰实验指南》或相关国际、国内有关体外诊断产品性能评估的文件进行特异性评估。 6.1交叉反应:易产生交叉反应的其他物质的验证情况,如与其他维生素D代谢产物的交叉反应情况。应至少验证与1,25(OH)2D2、1,25(OH)2D3的交叉反应情况。 6.2干扰物质:样本中常见干扰物质对检测结果的影响,如内源性干扰物(胆红素、血红蛋白、甘油三酯、总蛋白等)、类风湿因子(Rheumatoid Factor,RF)、抗核抗体(Antinuclear Antibody,ANA)、人抗鼠抗体(Human Antimouse Antibody,HAMA)等干扰因子的研究。 干扰物浓度的分布应覆盖人体样本生理及病理状态下可能出现的物质浓度。建议将研究结果在说明书中进行说明。如无法获得含有高浓度干扰物质的样本,可采用纯品物质分别添加到健康人样本、参考区间附近样本、中浓度值样本中的方式进行验证。方法为对模拟添加样本分别进行验证,样本量选择应体现一定的统计学意义,说明样本的制备方法及干扰试验的评价标准,确定可接受的干扰物质极限浓度,结果应量化表示,禁用轻度、严重的模糊表述。被测物浓度至少应包括其参考值的浓度。 6.3药物干扰的研究可根据需要由申请人选择是否进行或选择何种药物及其浓度进行。 6.4抗凝剂的干扰:如果申报试剂适用样本类型包括血浆样本,应采用各种适用抗凝剂抗凝的血浆样本分别与血清样本进行对比试验研究。方法为对比线性范围内的同一病人的血清和血浆样本(应包含参考值以及低值浓度样本的检测),以验证申报试剂对于血清和血浆样本检测结果的一致性。样本量选择应体现一定的统计学意义,说明样本的制备方法及干扰实验的评价标准,确定可接受的干扰物质极限浓度。 7.可报告范围(如适用) 可报告范围包括可报告低限与可报告高限。低值样本即将待测样本进行稀释,产生接近于方法线性范围低限浓度水平的样本,一般为5个浓度水平,浓度水平间隔应小于线性范围低限的10%,重复测定10次,选取CV值等于或小于可接受界值的最低浓度水平作为可报告范围低限。高值样本即选取含被测物的高值样本进行稀释,使其接近于线性范围的上1/3区域内,并记录稀释倍数。至少选用三个高浓度样本,稀释倍数应为方法性能标明的最大稀释倍数、并适当增加或减小稀释比例,重复测定3次,试验过程应明确稀释液类型,注意基质效应影响,必要时应提供基质效应研究有关的资料。选取还原浓度与理论浓度的偏差(%)等于或小于方法标示CV值时的最大稀释倍数为方法推荐的最大稀释倍数,方法线性范围的上限与最大稀释倍数的乘积为该方法可报告范围的高限。 8.其他需注意问题 8.1 对于适用多个机型的产品,应提供产品说明书【适用机型】项中所列的所有型号仪器的性能评估资料。对于某些性能,可采用主机型进行充分的性能研究,其他机型采用验证的试验方案进行。但对于主机型的判断,应有充分依据。 8.2 如有多个包装规格,需要对不同包装规格之间的差异进行分析或验证,如不同包装规格产品间存在性能差异,需要提交采用每个包装规格产品进行分析性能评估的资料。如不同包装规格之间不存在性能差异,企业需要提交包装规格之间不存在性能差异的分析,可仅选择代表规格进行分析性能评估。 8.3 针对技术要求中所列性能指标,应当对多批(至少三批)产品进行分析评估,并对结果进行统计分析。其他性能指标的评估申请人可结合自身产品特点确定评估批次/数量。 8.4 校准品溯源及质控品赋值(如适用) 应参照GB/T 21415—2008《体外诊断医疗器械
生物样品中量的测量 校准品和控制物质赋值的计量学溯源性》的要求,提供企业(工作)校准品及试剂盒配套校准品定值及不确定度的研究资料,提供质控品赋值及其靶值范围确定的研究资料。 8.5校准品及质控品性能评估(如适用) 如注册单元中包含校准品或质控品,建议应对其性能进行评估应至少包含外观、装量、准确度、均一性。 8.6生物安全性(如适用) 试剂盒如含人源性成分,至少使用经过国家批准合格的人类免疫缺陷病毒抗体诊断试剂盒、丙型肝炎病毒抗体诊断试剂盒、乙型肝炎病毒表面抗原诊断试剂盒、梅毒螺旋抗体诊断试剂盒,对该申报试剂盒(包括校准品、质控品)进行检测,检测结果应均为阴性。 (五)参考区间确定资料 应提交建立/验证参考区间所采用样本来源、详细的试验资料、统计方法等。明确参考人群的纳入、排除标准,离群值的处理,考虑不同年龄、性别、生活习惯、地域等因素,尽可能考虑样本来源的多样性、代表性,样本例数应符合统计学要求。 建议申请人根据临床实际情况,参考《WS/T 402-2012临床实验室检验项目参考区间的制定》或其他国际国内权威文件来确定申报试剂的参考区间。
|