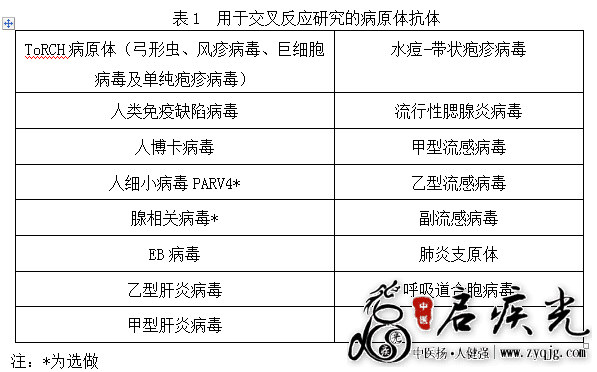
4.分析特异性 4.1交叉反应 4.1.1用于B19病毒交叉反应验证的病原体种类主要考虑以下几方面:抗原结构具有同源性(如在分类学上的近缘病毒)、易引起相同或相似的临床症状、易并发感染的其他微生物(见表1),以及高浓度病原体特异性IgG抗体与特异性IgM抗体的交叉反应验证。
4.1.2如果检测试剂采用基因重组抗原,应增加对重组基因导入微生物特异性抗体的交叉反应评价。例如,如果采用大肠杆菌作为宿主菌,建议考虑大肠杆菌自身蛋白或载体骨架编码的蛋白产生的抗体与被测物之间可能产生的交叉反应。 其中用于交叉反应研究用的抗体类型应与待测目的抗体类型相同,如申报两种抗体类型,用于交叉反应研究的病原体的IgM抗体、IgG抗体应分别进行验证。应提交所有用于交叉反应验证的病原体抗体样本来源、阴阳性、种属确认等相关信息。
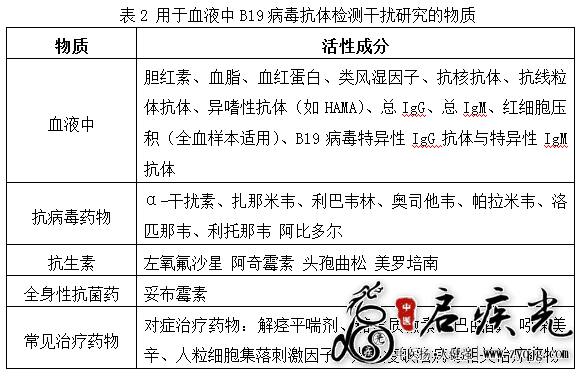
4.2干扰实验 4.2.1内源性及外源性干扰 应对样本中可能存在的内源性及外源性干扰物质(见表2)进行研究。建议对B19病毒抗体弱阳性、阳性的临床样本,使用潜在最大浓度的干扰物质分别进行添加,确定是否产生干扰。如有干扰,梯度稀释干扰物并进一步确定可接受的干扰物质的最高浓度水平,或产生干扰的浓度水平。应使用多份临床样本,每个样本重复检测不少于3次。申请人应描述干扰物质的种类,说明样本的制备方法及待测物的水平,以及不产生干扰的验收标准。 4.2.2抗凝剂的干扰 如果试剂盒适用样本类型包括血浆和/或全血样本,可采用一定数量血清、血浆、全血同源样本进行对比实验研究,以验证不同抗凝剂的适用性。 4.2.3 B19病毒IgM抗体检测特异性验证 对于IgM抗体检测试剂,可采用对至少10份含有B19病毒特异性IgM抗体的样本进行IgM破坏实验研究,方法为采用特定的化学制剂(如2-巯基乙醇或二硫苏糖醇)处理样本后,重新进行检测,IgM检测结果应为阴性;或者选用其他合理的方法进行验证。 5.精密度 具体试验方法可以参考相关的美国临床实验室标准化协会批准指南(CLSI-EP)文件或国内有关体外诊断产品性能评估的文件。企业应对每项精密度指标的验收标准做出合理要求,如标准差或变异系数的范围等。 5.1影响精密度的条件包括:操作者、测量仪器、测量程序、试剂批次、校准(校准品批次,校准周期)、运行、时间、地点、环境条件等。除申报试剂本身外,还应对以上可能影响检测精密度的主要变量进行验证。 5.2设定合理的精密度评价周期。 5.3用于精密度评价的样本应使用临床样本。至少包括三个水平:阴性、弱阳性以及中等阳性的样本。 6.钩状(HOOK)效应 建议采用高滴度的B19病毒抗体阳性临床样本进行梯度稀释后分别检测,每个梯度重复3至5次,将显色深度或检测值随滴度升高反而变浅或降低时的滴度作为出现钩状效应时B19病毒抗体的最低滴度。 7.企业参考品的验证(如适用): 根据主要原材料研究资料中企业参考品的设置情况,采用三批产品对企业参考品进行检验并提供详细的实验数据。 阳性判断值反映区分受试者目标状态的特征。申请人应考虑不同地理区域流行病学背景以及产生不同强度免疫应答反应的人群(如免疫缺陷/抑制人群)与正常人群之间的差异,选择具有代表性的样本建立阳性判断值,注意应纳入一定数量的弱阳性样本。 建议采用受试者工作特征曲线(ROC)的方法确定阳性判断值。如采用其他研究方法,应说明其合理性。若试验结果存在灰区,建议申请人从临床意义的角度出发,合理设定灰区范围并提供灰区的确定资料。 试验所用样本来源应考虑到年龄(成人或儿童)、不同的感染阶段(早期和晚期)和生理状态(是否妊娠)等因素的影响。另外,建议申请人考虑建立阳性判断值时使用的受试者样本对于目标人群的代表性,通过临床试验进一步确认阳性判断值的准确性。 (六)稳定性研究资料 稳定性研究资料主要涉及两部分内容,申报试剂的稳定性和适用样本的稳定性研究。前者主要包括实时稳定性、运输稳定性、开瓶稳定性(如涉及)及冻融稳定性(如涉及)等研究,申请人可根据实际需要选择合理的稳定性研究方案。稳定性研究资料应包括研究方法的确定依据、具体的实施方案、详细的研究数据以及结论。对于实时稳定性研究,应提供至少三批样品在实际储存条件下保存至成品有效期后的研究资料。 样本稳定性一般包括样本各种实际运输及储存(常温、冷藏和冷冻)条件下的保存期限验证,以确认样本的保存条件及效期(短期、长期)。需要冷冻保存的样本同时应对冻融次数进行合理验证。某些用于防腐、冷冻用途或起稳定保护作用的添加剂可能会对检测造成影响,如涉及,请对该添加剂的影响进行合理验证。 试剂稳定性和样本稳定性两部分内容的研究结果应在说明书【储存条件及有效期】和【样本要求】两项中进行详细说明。 (七)临床试验资料 临床试验的开展、方案的制定以及报告的撰写等均应符合相关法规及《体外诊断试剂临床试验技术指导原则》(原国家食品药品监督管理总局通告2014年第16号)的要求,如相关法规、文件有更新,临床试验应符合更新后的要求。 1.试验方法 临床试验可采用试验用体外诊断试剂与临床普遍认为质量较好的已上市同类产品进行比较研究试验,证明两者等效,从而间接证明试验用体外诊断试剂临床性能满足预期用途的要求。对比试剂在预期用途、适用人群、样本类型、检测性能等方面应与试验用体外诊断试剂具有较好的可比性。临床试验方案中应针对对比试剂的选择及依据进行详细描述。 对于B19病毒IgM检测试剂,申请人还应选择感染急性期患者采集的样本进行申报试剂与感染急性期判断的方法的一致性研究,以评价申报试剂的检测性能。用于评价的样本可选择处于感染急性期患者的采集样本(不少于30例);或者选择商业血清转化盘(不少于5套)。可采用的用于感染急性期判断的方法,例如:B19病毒特异性IgG抗体的血清学转换(动态监测2份或以上的血清IgG,恢复期与急性期比较IgG呈4倍以上升高)、核酸检测等方法,对于B19病毒感染急性期的判断应密切结合患者的临床诊断综合进行。 2.受试者选择及样本收集 临床试验方案中应根据试验用体外诊断试剂的预期用途、适用人群和检测要求等合理确定临床试验受试者选择要求,包括:受试者入组/排除标准、样本收集的前瞻性和回顾性设计等,并在临床试验过程中严格遵循。 临床试验的入组人群应为产品的预期适用人群,包括各种可能接受B19病毒感染检查的人群,例如具有疑似B19病毒感染症状的人群。 入组人群应包含不同年龄段、不同性别人群,以及一定数量的孕妇、免疫功能受损、接受免疫抑制治疗的人群,应尽量覆盖各类适用人群。 建议临床试验采用前瞻性收集的样本进行研究,同时,在严格控制偏倚的前提下,允许入组部分符合要求的回顾性样本。 另外,建议根据流行病学证据纳入不同地区的患者/人群,以研究本产品的临床检出能力。 3.临床试验机构数量及要求 该类产品临床试验应在三家及以上符合要求的临床试验机构开展。申办者应根据产品特点及其预期用途,综合流行病学背景,选择具有一定地域代表性的机构开展临床试验。原则上应具有相关学科的优势,实验操作人员有足够的时间熟悉检测系统的各环节,熟悉评价方案。
|